

A | B | C | D | E | F | G | H | CH | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | X | Y | Z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
1,3-dipolární cykloadice je chemická reakce mezi 1,3-dipólem a dipolarofilní sloučeninou, při níž vzniká pětičlenný cyklus. První takové reakce byly provedeny na přelomu 19. a 20. století, krátce po objevu 1,3-dipólů. Mechanismus a využití těchto reakcí v organické syntéze byly zjištěny v 60. letech 20. století, a to hlavně díky pracím Rolfa Huisgena.[1] Reakce tak někdy bývá označována jako Huisgenova cykloadice (tento název se používá zejména pro 1,3-dipolární cykloadiční reakce organického azidu s alkynem za vzniku 1,2,3-triazolu). 1,3-dipolární cykloadice je významnou metodou regio- a stereoselektivní přípravy pětičlenných heterocyklů a jejich acylových derivátů s otevřenými řetězci.

Mechanismus
Původně byly navrženy dva mechanismy 1,3-dipolární cykloadice: prvním je pericyklický cykloadiční mechanismus, navržený Rolfem Huisgenem;[2] druhým pak postupný mechanismus zahrnující biradikálový meziprodukt, jejž navrhl R. Firestone.[3] Po mnoha debatách je nyní víceméně přijímán první mechanismus,[4] kde 1,3-dipól reaguje s dipolarofilem ve spojené, často asynchronní, reakci a symetrií umožněné π4s + π2s produkty přes šestielektronový aromatický meziprodukt. Existuje ovšem několik příkladů postupného mechanismu nekatalytických 1,3-dipolárních cykloadičných reakcí u thiokarbonylylidů[5] a nitriloxidů[6]

Pericyklický mechanismus
Huisgen zkoumal řadu cykloadičních reakcí 1,3-dipolárních diazosloučenin s nejrůznějšími dipolarofilními alkeny.[2] Následující zjištění podporují spojený pericyklický mechanismus a vyvracejí postupný biradikálový nebo polární mechanismus:
- Vliv substituentů: Různé substituenty na dipólu nevykazují výrazný vliv na rychlost cykloadice, takže při reakci nevzniká nábojem izolovaný meziprodukt.
- Vliv rozpouštědel: Polarita použitého rozpouštědla jen málo ovlivňuje rychlost cykloadiční reakce, což je v souladu s pericyklickým mechanismem, kde polarita nezpůsobuje výrazné změny při přeměně reaktantů na přechodný stav.
- Stereochemie: 1,3-dipolární cykloadice jsou vždy stereospecifické s ohledem na dipolarofil (například z cis-alkenů vždy vznikají syn-produkty), což opět podporuje pericyklický mechanismus, kde se dvě vazby sigma tvoří současně.
- Termodynamické parametry: 1,3-dipolární cykloadice mají neobvykle vysoké záporné hodnoty entropie aktivace, podobně jako Dielsova–Alderova reakce, což naznačuje, že je přechodný stav velmi uspořádaný, a rovněž souhlasí s pericyklickým mechanismem.
1,3-dipól

1,3-dipól je organická molekula, která se může vyskytovat ve zwitteriontové oktetové/sextetové struktuře allylového i propargylového/allenylového typu. Oba tyto druhy 1,3-dipólů sdílejí čtyři elektrony v π-systému nad třemi atomy. Allylový typ je zakřivený zatímco propargylový/allenylový typ má lineární molekulární geometrii.[7] Jsou známy i 1,3-dipóly obsahující prvky vyšších řad periodické tabulky, jako jsou síra a fosfor, ovšem ty nejsou tak často využívány.
Rezonanční struktury lze zobrazit jako delokalizované kladné i záporné náboje na obou koncích dipólu (viz následující schéma). Přesnější metody pro popis rozdělení elektronů u 1,3-dipólů, jako například měření dipólového momentu, jsou založeny na experimentálních či teoretických datech[8] či výpočtech.[9] Například diazomethan má největší záporný náboj na koncovém atomu dusíku, zatímco azoimid má největší záporný náboj na prostředním atomu dusíku.

V souvislosti s tím mohou konce molekul 1,3-dipólů reagovat s nukleofilními i elektrofilními činidly najednou. Nukleofilnost a elektrofilnost každého konce lze zjistit využitím hraničních molekulových orbitalů, které lze získat s použitím výpočetní techniky. Obecně lze říci, že stom s největším orbitalovým koeficientem v rámci HOMO funguje jako nukleofil, zatímco atom v LUMO funguje jako elektrofil. Nejvíce nukleofilním atomem je obvykle, ne však pokaždé, ten, na němž je největší elektronová hustota.[10][11][12] U 1,3-dipolárních cykloadicí je identita páru dipól-dipolarofil určena tím, zda převažuje HOMO nebo LUMO charakter dipólu.
Dipolarofil
Nejčastěji používanými dipolarofily jsou alkeny a alkyny. Dipolarofily obsahující heteroatomy jako jsou karbonylové sloučeniny a iminy mohou také podstoupit 1,3-dipolární cykloadici. Dalšími příklady dipolarofilů jsou fullereny a nanotrubice, u nichž může proběhnout 1,3-dipolární cykloadice s azomethinylidy v Pratově reakci.
Stereospecifita
1,3-dipolární cykloadice často vyústí v „zadržení konfigurace“ s ohledem na 1,3-dipól a dipolarofil. Vysoká stereospecifita silně podporuje spojený reakční mechanismus oproti postupnému, při němž by byla nízká až nulová.
Vliv dipolarofilu
cis-substituenty na dipolarofilním alkenu vytváří cis- a trans-substituenty vytváří trans strukturu výsledné sloučeniny s pětičlenným cyklem.[13]

Vliv dipólu
Stereochemie dipólu obecně není objektem velkého zájmu, jelikož pouze několik dipólů může vytvořit stereogenní centra a rezonanční struktury umožňují rotaci vazby, čímž dochází k narušení stereochemie. Při studii azomethinylidů bylo však ověřeno, že stereospecifitu 1,3-dipolární cykloadice ovlivňují i dipóly. Diastereomerně čisté azomethinylidy se připraví elektrocyklickým otevřením cyklu aziridinů a následně se okamžitě zachytí silnými dipolarofily dříve, než může dojít k rotacím vazeb (viz níže uvedený obrázek).[14][15] Při použití slabších dipolarofilů mají vazby na dipólu dostatek času k rotaci a dojde tak k nespárované stereospecifitě cykloadice.
Tyto výsledky potvrzují, že 1,3-dipolární cykloadice je stereospecifická a že stereospecifitu ovlivňují 1,3-dipól i dipolarofil.
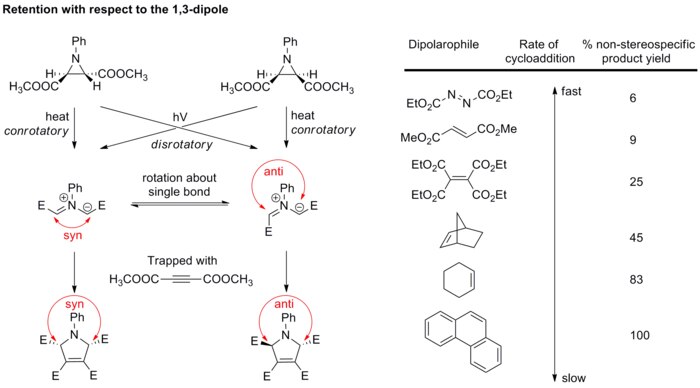
Diastereospecifita
Když se během reakce vytvoří dvě nebo více chirálních center, lze zíksat diastereomerní přechodné stavy a produkty. V Dielsově–Alderově cykloadici je obvykle pozorována endo diastereoselektivita, což je způsobeno sekundárními orbitalovými interakcemi. U 1,3-dipolárních cykloadicí ovšem diastereoselektivitu ovlivňují dvě síly: přitažlivé π interakce (podobné sekundárním orbitalovým interakcím u Dielsovy–Alderovy reakce) a odpudivé sterické interakce. Tato působení se často navzájem vyruší, což vede k nízké diastereoselektivitě 1,3-dipolárních cykloadicí.
Příklady substrátem řízené diastereoselektivních 1,3-dipolárních cykloadicí jsou zobrazeny níže. Prvním je reakce benzonitrilu N-benzylidu a methylakrylátu. V přechodných stavech se fenylová a methylesterová skupina spojí tak, že dojde k cis substituci za vzniku výsledného pyrrolinového produktu. Upřednostňovaná π interakce vyváží vzájemné sterické odpuzování fenylové a methylesterové skupiny.[16] Druhým je reakce nitronu a dihydrofuranu. Dosažením exo selektivity se omezí sterické odpuzování.[17] Poslední případ představuje vnitromolekulární reakce azomethinového ylidu s alkenem. Diastereoselektivita je ovládána tvorbou méně narušovaného cis systému spojených cyklů.[18]

Řízená 1,3-dipolární cykloadice
Cykloadici lze ovládat a dosáhnout tak diastereoselektivní reakce. Například kovové ionty mohou být chelatovány dipolarofilem a dipólem a řídit tak reakci selektivně. Níže uvedený obrázek zobrazuje adici nitriloxidu na enantiomerně čistý allylalkohol za přítomnosti hořečnatých kationtů. Nejstabilnější konformace alkenu je ta, u níž se hydtroxylová skupina nachází nad rovinou alkenu. Hořčík je následně chelatován hydroxylovou skupinou a kyslíkovým atomem nitriloxidu. Cykloadice tedy probíhá selektivně.[19]
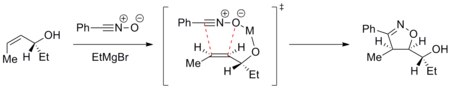
Metody diastereoselekce se využívají například při syntéze epothilonů.[20]

Regioselektivita
U asymetrických párů dipól-dipolarofil mohou vzniknout dva regioizomerní produkty. Regioselektivitu 1,3-dipolárních cykloadicí ovlivňují jak elektronové/stereoelektronové jevy, tak i sterické faktory.[21]
Elektronové/stereoelektronové jevy
Převažující interakcí během 1,3-dipolárních cykloadicí je interakce mezi největším HOMO a největším LUMO. Regioselektivita je tak řízena atomy s největšími HOMO a LUMO orbitalovými koeficienty.[22][23]
Jako příklad lze uvést cykloadici diatzomethanu na tři různé dipolarofily: methylakrylát, styren a methylcinnamát. Uhlíkový atom diazomethanu má největší HOMO, zatímco koncové alkenové uhlíky methylakrylátu a styrenu mají největší LUMO; cykloadice tedy probíhá regioselektivně na pozici C3. U methylcinnamátu oba substituenty (fenylová a methoxykarbonylová skupina) „soutěží“ v odtahování elektronů. Převládne vliv karboxylu, díky čemuž je nejelektrofilnější β uhlík. Karboxylová skupina se tak regioselektivně připojuje na pozici C3 a fenylová skupina na pozici C4.

Sterické efekty
Sterické efekty mohou výše zmíněné elektronové jevy podporovat, ale mohou též působit proti nim. V některých případech sterické efekty zcela převáží nad elektronovými a vzniká tak výhradně opačný regioizomer;[24] například diazomethan většinou reakcí s methylakrylátem vytváří 3-karboxylpyrazolin. Při zesilování sterických jevů ovšem začíná vznikat rovněž izomerní 4-karboxylpyrazolin, poměr množství těchto izomerů závisí na intenzitě sterických jevů. Zvětšení velikosti substituentů od atomu vodíku k terc-butylu zcela obrátí regioselektivitu ze 100 % 3-karboxylové na 100 % 4-karboxylové substituce.[25][26]

Využití v syntéze
1,3-dipolární cykloadice jsou významnými postupy syntézy řady důležitých pětičlenných heterocyklů jako jsou například triazoly, furany, isoxazoly a pyrrolidiny. Některé cykloadukty mohou být štěpeny za vzniku lineárního řetězce, což poskytuje další možný způsob přípravy alifatických sloučenin. Tyto reakce jsou velmi významné rovněž díky své stereospecifitě, diastereoselektivitě a regioselektivitě.
Reakce s nitriloxidy
1,3-dipolární cykloadice s nitriloxidy je často používanou maskovanou aldolovou reakcí. Cykloadiční reakcí nitriloxidu s alkenem vzniká cyklický isoxazolinový produkt, zatímco reakce s alkynem poskytuje isoxazol. Isoxazoliny i isoxazoly mohou být rozštěpeny hydrogenací za vzniku β-hydroxykarbonylových sloučenin aldolového typu nebo β-dikarbonylových sloučenin Claisenova typu.
Nitriloxidová-alkynová cykloadice následovaná hydrogenací byla využita při syntéze miyakolidu, jak je uvedeno na následujícím obrázku:[27]

Karbonylylidy
1,3-dipolární cykloadiční reakce byly vyvinuty jako výborné metody k přípravě komplexních cyklických řetězců a molekul pro lékařské, biologické a mechanistické výzkumy. Konkrétně cykloadice se zahrnutím karbonylylidů byly široce využity na přípravu molekul s pětičlennými cykly obsahujícími kyslík.[28]
Příprava karbonylylidů pro 1,3-dipolární cykloadice
Ylidy jsou považovány za kladně nabité heteroatomy spojené se záporně nabitými uhlíkovými atomy, patří k nim též ylidy sulfonia, thiokarbonylu, oxonia, dusíku a karbonylu.[29] Karbonylylidy, důležité meziprodukty při přípravě pětičlenných cyklů obsahujících kyslík, lze připravtit několika způsoby.
Příprava karbonylylidů z derivátů diazomethanu fotokatalýzou
Jedna z prvních metod, z roku 1983, syntézy karbonylylidů v sobě zahrnuje fotokatalýzu.[30] Fotolýzou diazotetrakis(trifluormethyl)cyklopentadienu (DTTC) za přítomnosti tetramethylmočoviny lze připravit karbonylylid vnitromolekulární nukleofilní reakcí a následnou aromatizací zbylé DTTC.[30] Produkt byl izolován a poté analyzován rentgenovou krystalografií kvůli stabilitě získané díky aromaticitě, trifluormethylovým skupinám snižujícím a dimethylaminovým skupinám zvyšujícím elektronovou hustotu. Stabilní karbonylylidové dipóly mohou být následně využity k cykloadičním reakcím s dipolarofily.

Další příklad syntézy karbonylylidů byl popsán roku 1986.[31] Dideuteriodiazomethan byl fotolyticky rozložen nza přítomnosti formaldehydu a vznikl dideuterioformaldehydový karbonylylid.

Příprava karbonylylidů z hydroxypyronů přesunem protonů
Karbonylylidy lze připravit kysele katalyzovanou reakcí z hydroxy-3-pyronů za nepřítomnosti kovového katalyzátoru.[32] Na začátku dochází k tautomerizaci, následuje eliminace odcházející skupiny, což vede k aromatizaci pyronového kruhu a vzniku karbonylylidu; z něj pak lze cykloadicí vytvořit oxacyklus. Tento způsob se nepoužívá tak často kvůli požadavkům na pyronové sloučeniny a také kvůli omezené využitelnosti.

5-hydroxy-4-pyrony lze též použít na přípravu karbonylylidů vnitromolekulárním přesunem vodíkového protonu.[33] Po provedení reakce může vzniklý karbonylylid reagovat s dipolarofily za vzniku kyslíkatých heterocyklů.

Příprava α halogenkarbonylylidů z dihalogenkarbenů
K tvorbě karbonylylidů lze využít i dihalogenkarbeny, přičemž se využívá schopnost těchto sloučenin snižovat elektronovou hustotu.[34][35][36] Jako zdroj dichlorkarbenů lze použít fenyl(trichlormethyl)rtuť a jako zdroj dibromkarbenů se dá použít fenyl(tribrommethyl)rtuť. Karbonylylid je možné získat reakcí dihalogenkarbenů s ketony nebo aldehydy. Při přípravě α halogenkarbonylylidů ovšem může dojít k nežádoucí ztrátě molekuly oxidu uhelnatého a následnému vzniku deoxygenovaného produktu.

Příprava karbonylylidů z derivátů diazomethanu za katalýzy sloučeninami kovů
Jedním z možných způsobů přípravy karbonylylidů jsou sloučeninami kovů katalyzované reakce α diazokarbonylových sloučenin, jako katalyzátor se nejčastěji používají mědné nebo rhodné sloučeniny.[37] Po uvolnění plynného dusíku a přeměně na metalokarben dochází ke vnitromolekulární reakci, jejímž produktem je karbonylylid; tetn poté cykloadiční reakcí s alkenovým nebo alkynovým dipolarofilem vytvoří pětičlenný cyklus obsahující kyslíkový atom. K nejčastějším katalyzátorům takovýchto reakcí patří Rh2(OAc)4 a Cu(acac)2.[38][39]

Azomethinylidy
1,3-dipolární cykloadiční reakcí azomethinylidu s alkenem vzniká sloučenina s azacyklickou strukturou jako například pyrrolidin. Tento postup se mimo jiné využívá při syntéze spirotryprostatinu A.[40]
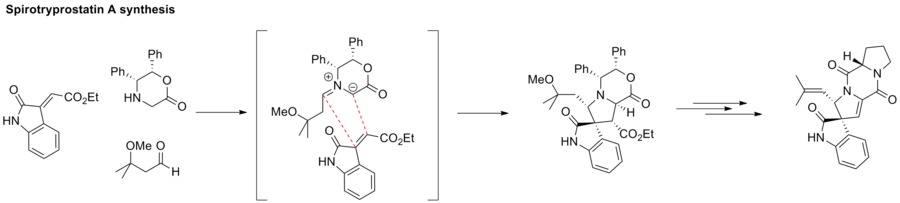
Biologická využití
1,3-dipolární cykloadice mezi organickými azidy a koncovými alkyny (například azido-alkynová Huisgenova cykloadice) se využívá v biokonjugaci.
Katalýza měďnými sloučeninamieditovat | editovat zdroj
Huisgenova reakce obecně při nízkých teplotách neprobíhá příliš rychle. Byla vyvinuta její varianta katalyzovaná měďnými sloučeninami Cu+, která probíhá velmi rychle i za mírných, například fyziologických, podmínek (neutrální pH, pokojová teplota a vodný roztok).[41][42] Tato reakce je také bioortogonální: azidy i alkyny se v biologických systémech obvykle nevyskytují a tak mohou být provedeny chemoselektivně i v rámci buňky. Tyto látky rovněž nereagují s ostatními funkčními skupinami vyskytujícími se v přírodě, a tak biologické systémy nenarušují. Měďné sloučeniny jsou cytotoxické, ovšem bylo vyvinuto mnoho ligandů, které snižují cytotoxicitu a zvyšují rychlost reakce, což umožňuje její využití při in vivo studiích.[43]

Bylo například popsáno metabolické zařazení do glykanů v buněčných membránách u azidy funkcionalizovaných sacharidů a jejich následné značkování komplexy fluoroforů s alkyny. Fluorescenčně značkovaná buněčná membrána může být zobrazena pomocí fluorescenčního mikroskopu.[44]

Napětím podporovaná cykloadiceeditovat | editovat zdroj
Aby zamezili toxickým účinkům jednomocné mědi, vyvinuli Carolyn Bertozzi et al. napětím podporovanou azido-alkynovou cykloadiční reakci (SPAAC) mezi organickým azidem a derivátem cyklooktynu. Úhlové pokřivení cyklooktynu umožňuje urychlit reakci, díky čemuž ji lze prakticky provést za fyziologických podmínek i bez použití katalyzátoru.[45]

Alice Ting et al. zabudovali azidofunkcionalitu do určitých bílkovin na povrchu buněk za použití ligázového enzymu. Tyto bílkoviny byly následně označkovány cyklooktyno-fluoroforovým komplexem za vzniku fluorescenčně zbarveného proteinu.[46]

Referenceeditovat | editovat zdroj
V tomto článku byl použit překlad textu z článku 1,3-Dipolar cycloaddition na anglické Wikipedii.
- ↑ HUISGEN, Rolf. 1.3-Dipolare Cycloadditionen Ruckschau und Ausblick.. Angewandte Chemie. 1963, roč. 75, s. 604–637. Dostupné online abstract. (anglicky)
- ↑ a b HUISGEN, Rolf. Kinetics and Mechanism of 1,3-Dipolar Cycloadditions. Angewandte Chemie International Edition. November 1963, roč. 2, čís. 11, s. 633–645. Dostupné v archivu pořízeném dne 2012-12-11. DOI 10.1002/anie.196306331. (anglicky)
- ↑ FIRESTONE, R. Mechanism of 1,3-dipolar cycloadditions. Journal of Organic Chemistry. 1968, roč. 33, s. 2285–2290. Dostupné online. DOI 10.1021/jo01270a023. (anglicky)
- ↑ HUISGEN, Rolf. 1,3-Dipolar cycloadditions. 76. Concerted nature of 1,3-dipolar cycloadditions and the question of diradical intermediates. Journal of Organic Chemistry. 1976, roč. 41, s. 403–419. Dostupné online. DOI 10.1021/jo00865a001. (anglicky)
- ↑ MLOSTON, G.; LANGHALS, E.; HUISGEN, Rolf. First Two-Step 1,2-Dipolar Cycloadditons: Nonstereospecificity. J. Am. Chem. Soc.. 1986, roč. 108, s. 6401–66402. Dostupné online. DOI 10.1021/ja00280a053. (anglicky)
- ↑ SEYYED AMIR, Siadati. An example of a stepwise mechanism for the catalyst-free 1,3-dipolar cycloaddition between a nitrile oxide and an electron rich alkene. Tetrahedron Letters. 2015, roč. 56, s. 4857–4863. Dostupné online. DOI 10.1016/j.tetlet.2015.06.048. (anglicky)
- ↑ HUISGEN, Rolf. 1,3-Dipolar Cycloadditions. Past and Future. Angewandte Chemie International Edition. 1963, roč. 2, s. 565–598. Dostupné online. DOI 10.1002/anie.196305651. (anglicky)
- ↑ COX, A; THOMAS, L; SHERIDAN, J. Microwave Spectra of Diazomethane and its Deutero Derivatives. Nature. 1958, roč. 181, čís. 4614, s. 1000–1001. Dostupné online. DOI 10.1038/1811000a0. Bibcode 1958Natur.181.1000C. (anglicky)
- ↑ HILBERTY, P; LEFORESTIER, C. Expansion of molecular orbital wave functions into valence bond wave functions. A simplified procedure.. Journal of the American Chemical Society. 1978, roč. 100, s. 2012–2017. Dostupné online. DOI 10.1021/ja00475a007. (anglicky)
- ↑ MCGARRITY, J.F.; PATAI, Saul. Basicity, acidity and hydrogen bonding. Diazonium and Diazo Groups. 1978, roč. 1, s. 179–230. Dostupné online. DOI 10.1002/9780470771549.ch6. (anglicky)
- ↑ BERNER, Daniel; MCGARRITY, John. Direct observation of the methyldiazonium ion in fluorosulfuric acid. Journal of the American Chemical Society. 1979, roč. 101, s. 3135–3136. Dostupné online. DOI 10.1021/ja00505a059. (anglicky)
- ↑ MULLER, Eugen; RUNDEL, Wolfgans. Untersuchungen an Diazomethanen, VI. Mitteil.: Umsetzung von Diazoäthan mit Methyllithium. Chemische Berichte. 1956, roč. 89, s. 1065–1071. Dostupné online. DOI 10.1002/cber.19560890436. (anglicky)
- ↑ BIHLMAIER, Werner; GEITTNER, Jochen; HUISGEN, Rolf; REISSIGP, Hans-Ulrich. The Stereospecificity of Diazomethane Cycloadditions. Heterocycles. 1978, roč. 10, s. 147–152. Dostupné online. DOI 10.3987/S-1978-01-0147. (anglicky) Archivováno 11. 6. 2019 na Wayback Machine.
- ↑ HUISGEN, Rolf; SCHEER, Wolfgang; HUBER, Helmut. Stereospecific Conversion of cis-trans Isomeric Aziridines to Open-Chain Azomethine Ylides. Journal of the American Chemical Society. 1967, roč. 89, s. 1753–1755. Dostupné online. DOI 10.1021/ja00983a052. (anglicky)
- ↑ DAHMEN, Alexander; HAMBERGER, Helmut; HUISGEN, Rolf; MARKOWSKI, Volker. Conrotatory ring opening of cyanostilbene oxides to carbonyl ylides. Journal of the Chemical Society D: Chemical Communications. 1971, čís. 19, s. 1192–1194. Dostupné online. DOI 10.1039/C29710001192. (anglicky)
- ↑ PADWA, Albert; SMOLANOFF, Joel. Photocycloaddition of arylazirenes with electron-deficient olefins. Journal of the American Chemical Society. 1971, roč. 93, s. 548–550. Dostupné online. DOI 10.1021/ja00731a056. (anglicky)
- ↑ IWASHITA, Takashi; KUSUMI, Takenori; KAKISAWA, Hiroshi. A Synthesis of dl-isoretronecanol. Chemistry Letters. 1979, čís. 11, s. 1337–1340. Dostupné online. DOI 10.1246/cl.1979.1337. (anglicky) Archivováno 4. 7. 2018 na Wayback Machine.
- ↑ WANG, Chia-Lin; RIPKA, William; CONFALONE, Pat. A short and stereospecific synthesis of (±)-α-lycorane. Tetrahedron Letters. 1984, roč. 25, s. 4613–4616. Dostupné online. DOI 10.1016/S0040-4039(01)91213-4. (anglicky)
- ↑ KANEMASA, Shuji. Metal-Assisted Stereocontrol of 1,3-Dipolar Cycloaddition Reactions. Synthesis Letters. 2002, roč. 2002, s. 1371–1387. Dostupné online. DOI 10.1055/s-2002-33506. (anglicky)
- ↑ BODE, Jeffrey; CARREIRA, Erick. Stereoselective Syntheses of Epothilones A and B via Directed Nitrile Oxide Cycloaddition.. Journal of the American Chemical Society. 2011, roč. 123, čís. 15, s. 3611–3612. Dostupné online. DOI 10.1021/ja0155635. PMID 11472140. (anglicky)
- ↑ Vsevolod V. Rostovtsev; Luke G. Green; Valery V. Fokin; K. Barry Sharpless. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective Ligation of Azides and Terminal Alkynes. Angewandte Chemie International Edition. 2002, roč. 41, čís. 14, s. 2596–22599. DOI 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. PMID 12203546. (anglicky)
- ↑ CARAMELLA, Pierluigi; HOUK, K.N. Geometries of nitrilium betaines. The clarification of apparently anomalous reactions of 1,3-dipoles. Journal of the American Chemical Society. 1976, roč. 98, s. 6397–6399. Dostupné online. DOI 10.1021/ja00436a062. (anglicky)
- ↑ CARAMELLA, Pierluigi; GANDOUR, Ruth W.; HALL, Janet A.; DEVILLE, Cynthia G. A derivation of the shapes and energies of the molecular orbitals of 1,3-dipoles. Geometry optimizations of these species by MINDO/2 and MINDO/3. Journal of the American Chemical Society. 1977, roč. 99, s. 385–392. Dostupné online. DOI 10.1021/ja00444a013. (anglicky)
- ↑ HUISGEN, Rolf. Kinetics and Mechanism of 1,3-Dipolar Cycloadditions. Angewandte Chemie International Edition. November 1963, roč. 2, čís. 11, s. 633–645. Dostupné v archivu pořízeném dne 2012-12-11. DOI 10.1002/anie.196306331. (anglicky)
- ↑ PADWA, Albert. 1,3-Dipolar Cycloaddition Chemistry. United States of America: Wiley-Interscience, 1983. ISBN 0-471-08364-X. S. 141–145.
- ↑ KOSZINOWSKI, J.. thesis. , 1980. Ph.D.. .
- ↑ EVANS, David; RIPIN, David; HALSTEAD, David; CAMPOS, Kevin. Synthesis and Absolute Stereochemical Assignment of (+)-Miyakolide. Journal of the American Chemical Society. 1999, roč. 121, s. 6816–6826. Dostupné online. DOI 10.1021/ja990789h. (anglicky)
- ↑ Synthetic Reactions of M=C and M=N Bonds: Ylide Formation, Rearrangement, and 1,3-Dipolar Cycloaddition; Hiyama, T. W., J., Ed.; Elsevier, 2007; Vol. 11.
- ↑ Padwa, A.; Hornbuckle, S. F. Ylide Formation from the Reaction of Carbenes and Carbenoids with Heteroatom Lone Pairs Chem Rev 1991, 91, 263.
- ↑ a b Janulis, E. P.; Arduengo, A. J. Structure of an Electronically Stabilized Carbonyl Ylide J Am Chem Soc 1983, 105, 5929.
- ↑ Prakash, G. K. S.; Ellis, R. W.; Felberg, J. D.; Olah, G. A. Formaldehyde 0-Methylide, CH2=O+-CH2: The Parent Carbonyl Ylide J Am Chem Soc 1986, 108, 1341.
- ↑ Sammes, P. G.; Street, L. J. Intra molecular Cyclo additions with Oxido pyrylium Ylides J. Chem. Soc., Chem. Commun. 1982, 1056.
- ↑ Garst, M. E.; McBride, B. J.; Douglass III, J. G. Intramolecular cycloadditions with 2-(ω-alkenyl)-5-hydroxy-4-pyrones Tetrahedron Lett. 1983, 24, 1675.
- ↑ Gisch, J. F.; Landgrebe, J. A. Dichlorocarbene from flash vacuum pyrolysis of trimethyl(trichloromethyl)silane. Possible observation of 1,1-dichloro-3-phenylcarbonyl ylide J Org Chem 1985, 50, 2050.
- ↑ Huan, Z. W.; Landgrebe, J. A.; Peterson, K. Dibromocarbonyl ylides: Deoxygenation of aldehydes and ketones by dibromocarbene J Org Chem 1983, 48, 4519.
- ↑ Martin, C. W.; Lund, P. R.; Rapp, E.; Landgrebe, J. A. Halogenated carbonyl ylides in the reactions of mercurial dihalocarbene precursors with substituted benzaldehydes J Org Chem 1978, 43, 1071.
- ↑ Hodgson, D. M.; Bruckl, T.; Glen, R.; Labande, A. H.; Selden, D. A.; Dossetter, A. G.; Redgrave, A. J. Catalytic enantioselective intermolecular cycloadditions of 2-diazo-3,6-diketoester-derived carbonyl ylides with alkene dipolarophiles Proceedings of the National Academy of Sciences of the United States of America 2004, 101, 5450.
- ↑ Padwa, A.; Hertzog, D. L.; Nadler, W. R. Intramolecular Cycloaddition of Isomunchnone Dipoles to Heteroaromatic π-Systems J Org Chem 1994, 59, 7072.
- ↑ Hamaguchi, M.; Ibata, T. New Type of Mesoionic System. 1,3-Dipolar Cycloaddition of Isomunchnon With Ethylenic Compounds Archivováno 11. 4. 2020 na Wayback Machine. Chem Lett 1975, 499.
- ↑ ONISHI, Tomoyuki; SEBAHAR, Paul; WILLIAMS, Robert. Concise, Asymmetric Total Synthesis of Spirotryprostatin A. Organic Letters. 2003, roč. 5, s. 3135–3137. Dostupné online. DOI 10.1021/ol0351910. PMID 12917000. (anglicky)
- ↑ TORNOE, Christian; CHRISTENSEN, Caspar; MELDAL, Morten. Peptidotriazoles on Solid Phase: 1,2,3-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. Journal of Organic Chemistry. 2002, roč. 67, čís. 9, s. 3057–3064. DOI 10.1021/jo011148j. PMID 11975567. (anglicky)
- ↑ ROSTOVTSEV, Vsevolod; GREEN, Luke; FOKIN, Valery; SHARPLESS, Barry K. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective Ligation of Azides and Terminal Alkynes. Angewandte Chemie International Edition. 2002, roč. 41, s. 2596–2599. DOI 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. PMID 12203546. (anglicky)
- ↑ BESANCENEY-WEBLER, Christen; JIANG, Hao; ZHENG, Tianqing; FENG, Lei. Increasing the Efficacy of Bioorthogonal Click Reactions for Bioconjugation: A Comparative Study. Angewandte Chemie International Edition. 2011, roč. 50, s. 8051–8056. Dostupné online. DOI 10.1002/anie.201101817. PMID 21761519. (anglicky)
- ↑ BREIDENBACH, Mark; GALLAGHER, Jennifer; KING, David; SMART, Brian. Targeted metabolic labeling of yeast N-glycans with unnatural sugars. Proceedings of the National Academy of Sciences of the United States of America. 2010, roč. 107, čís. 9, s. 3988–3993. Dostupné online. DOI 10.1073/pnas.0911247107. PMID 20142501. Bibcode 2010PNAS..107.3988B. (anglicky)
- ↑ AGARD, Nicholas; PRESCHER, Jennifer; BERTOZZI, Carolyn. A Strain-Promoted 3 + 2 Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. Journal of the American Chemical Society. 2004, roč. 126, s. 15046–15047. DOI 10.1021/ja044996f. PMID 15547999. (anglicky)
- ↑ FERNANDEZ-SUAREZ, Marta; BARUAH, Hemanta; MARTINEZ-HERNANDEZ, Laura; XIE, Kathleen. Redirecting lipoic acid ligase for cell surface protein labeling with small-molecule probes. Nature Biotechnology. 2007, roč. 25, s. 1483–1487. Dostupné online. DOI 10.1038/nbt1355. PMID 18059260. (anglicky)
Externí odkazyeditovat | editovat zdroj
 Obrázky, zvuky či videa k tématu 1,3-dipolární cykloadice na Wikimedia Commons
Obrázky, zvuky či videa k tématu 1,3-dipolární cykloadice na Wikimedia Commons
Text je dostupný za podmienok Creative Commons Attribution/Share-Alike License 3.0 Unported; prípadne za ďalších podmienok. Podrobnejšie informácie nájdete na stránke Podmienky použitia.
Antropológia
Aplikované vedy
Bibliometria
Dejiny vedy
Encyklopédie
Filozofia vedy
Forenzné vedy
Humanitné vedy
Knižničná veda
Kryogenika
Kryptológia
Kulturológia
Literárna veda
Medzidisciplinárne oblasti
Metódy kvantitatívnej analýzy
Metavedy
Metodika
Text je dostupný za podmienok Creative
Commons Attribution/Share-Alike License 3.0 Unported; prípadne za ďalších
podmienok.
Podrobnejšie informácie nájdete na stránke Podmienky
použitia.
www.astronomia.sk | www.biologia.sk | www.botanika.sk | www.dejiny.sk | www.economy.sk | www.elektrotechnika.sk | www.estetika.sk | www.farmakologia.sk | www.filozofia.sk | Fyzika | www.futurologia.sk | www.genetika.sk | www.chemia.sk | www.lingvistika.sk | www.politologia.sk | www.psychologia.sk | www.sexuologia.sk | www.sociologia.sk | www.veda.sk I www.zoologia.sk